


在地球生态系统中,细菌、真菌、古菌、病毒以及真核生物共同构成了生命的基础,并在多个层面上发挥着重要的作用。人类和其他大型生物的反应往往可以直接观察到,但看不见的微生物的作用和影响也同样重要。
在微生物研究中,一直面临着一个棘手的难题:想要准确识别环境样本中的微生物,却常常遇到技术和成本障碍。传统上,研究人员需要分别测序16S和18S两种核糖体RNA,这不仅增加了研究成本,还可能因为使用"通用"引物导致鉴定结果产生偏差。
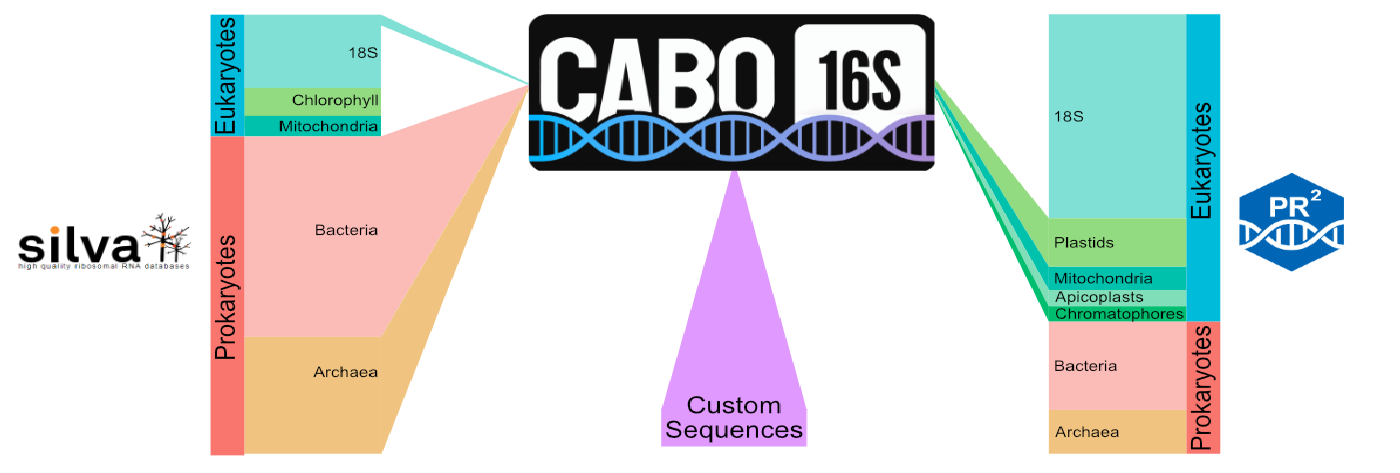
为解决这一问题,加州理工学院的研究团队在2024年10月创建了一个数据库——CABO-16S。这个数据库的独特之处在于将两个知名数据库(SILVA和PR2)中的微生物序列巧妙整合,让研究人员可以用更简单、更经济的方式获取微生物分类信息。
具体来说,CABO-16S将细菌、古细菌的序列与细胞器(如叶绿体)的16S rRNA序列结合,大大提高了微生物鉴定的准确性。特别是在海洋、湖泊和海草环境中,这个数据库在识别光合真核生物方面表现出色。
此外,CABO-16S还提供了一个灵活的框架,研究者可以添加专业的、尚未被其他数据库收录的微生物序列。本文以甲烷渗漏沉积物中的特定微生物为例,展示了如何通过添加精选序列来揭示微生物群落的细微差别。
这项创新不仅降低了研究成本,还为探索极端或独特环境中的微生物提供了新的可能性。
传统方法的不足
在过去20年中,小亚基(SSU)核糖体RNA(rRNA)的高通量测序用于研究陆地和海洋环境中的微生物生态学,并加深了对植物和动物微生物组的理解。通常16S rRNA基因用于原核生物鉴定,而优化的18S rRNA基因用于真核生物鉴定,ITS区域则被证明最适合真菌。
用单个PCR反应扩增原核和真核微生物是更为理想的,与使用单独引物进行16S和18S rRNA基因测序相比,可以将扩增子文库制备成本降低两到三倍。尽管一些“通用”引物 (515f/926r)可以扩增真核生物 18S rRNA 和 16S rRNA ,但同时准确分析来自 16S 和 18S rRNA 的真核生物和原核生物具有一定的难度。
◮ 真核生物被低估
首先,当试图扩增更广泛的目标群体时,引物与其模板之间的不匹配可能更常见,事实上,在模拟群落中,仅与反向引物的单个不匹配就会导致真核生物被低估3-8倍。
其次,18S序列通常比16S序列长160- 180bp,并且 PCR 和测序都偏向于较长的扩增子。天然样品可能含有较长的18S序列或较高比例的甲藻,这些甲藻往往存在错****配,这表明在一些天然样品中,515f/926r引物扩增真核生物可能被严重低估。
◮ 不能完全可靠地表示相对分类丰度
最后,在大多数细菌中,16S rRNA基因拷贝数在1-15之间,在大多数古细菌门中平均只有1个拷贝,而在浮游植物中,18S基因拷贝数可以在1-12,000之间变化。尽管18S基因计数可能与生物量显著相关,但它们不能可靠地用于表示相对分类丰度。
CABO-16S的优势
◮ 同时分析原核和真核生物更全面、更精准
CABO-16S作为一个整合16S rRNA序列的创新数据库,为微生物生态学研究提供了更全面和精确的分类工具。CABO-16S将细菌、古细菌的序列与细胞器(如叶绿体)的16S rRNA序列结合,特别适合需要同时分析原核生物和真核生物(尤其是光合真核生物)的环境样本研究,尤其是海洋、湖泊和沿海生态系统。
◮ 可根据研究需求添加自定义序列
研究者可以根据自身研究需求,向数据库添加自定义序列,但需谨慎操作,避免引入分类学不一致性。对于特定环境或复杂微生物群落的研究,建议结合基因组系统发育信息来验证16S rRNA分类结果。
在具体使用时由于数据库仍在持续更新,研究者应关注SILVA和PR2数据库的最新版本,并及时更新CABO-16S数据库,以获得更准确的微生物分类信息。
参考数据库集合
◮ 获取SILVA数据
为构建 CABO-16S 数据库,下载最新版本的 Silva(138.2)序列 (SILVA_138.2_SSURef_NR99_tax_silva.fasta.gz) 以及映射的分类法(taxmap_slv_ssu_ref_nr_138.2.txt.gz)和质量值 (SILVA_138.2_SSURef_Nr99.quality.gz)。
去除了针尾值 < 50 或比对质量值 < 75 的所有序列。去掉鉴定为叶绿体、线粒体和真核生物的序列,随机选择100个真核生物作为外群重新添加。真核生物外群的分类学仅保留门水平。
对原核生物分类学进行清理,特别是在物种水平,以删除基于生物宿主、样本收集、不明确的细菌分组或属重复(即“Genus sp.”)的命名方案。为了便于直接比较,使用相同的方法构建了一个简化的 SILVA 数据库,但保留了鉴定为叶绿体的序列。
◮ PR2数据库获取叶绿体、染色质等序列
将质体、顶质体、叶绿体和染色质序列从PR 2数据库(v 5.0.0)添加到CABO-16S数据库中,用R包“pr 2database”(pr2database.github.io/pr2database/articles/pr2database.html)获取。
为了与SILVA分类中的7个分类等级相匹配,从PR2序列中删除了超群和亚群的等级。
◮ 将SILVA数据与PR2数据相结合
最后,将从甲烷渗漏 Sanger 测序中获得的定制的16S rRNA序列与来自 SILVA 和 PR2 的数据相结合,形成 CABO-16S 数据集的基础。
CABO-16S和简化的SILVA 138.2训练集是根据DECIPHER的建议和IDTAXA算法制作的。简而言之,在使用LearnTasa函数进行三次迭代训练之前,将过采样组被随机子集化为100个序列。
Kmer长度设置为8nt,以匹配RDP和QIIME2默认值。注意,使用全长16S rRNA参考序列用于训练;截断扩增子窗口的可能会略微提高准确性,但代价是可能产生模糊性。因此,我们呈现全长序列并从全长序列进行比较,并将是否截断的选择留给用户。
基准测试数据集的分类
利用CABO-16S和SILVA-132.1对已发表的广泛来源16S rRNA序列进行分类比较,包括已知细菌分离株的模拟群落和环境样品。
对于所有比较的样品,使用古细菌/细菌引物(515 f/926 r)扩增16S rRNA基因的V4-V5区域,并在Illumina MiSeq平台上测序。在5个以上样本的环境数据集中,选择任意一组子样本进行分类比较。
对所有下载的原始序列进行了相同的处理,除了Needham和Fuhrman(2016)的数据,这些数据直接下载并使用了已经分析的OTU序列和观测矩阵。可重现的工作流程(github.com/emelissa3/CABO-16S commit 472d7fc)报告了用于从NCBI SRA上可用的原始FASTQ文件生成扩增子序列变体(ASVs)的全部细节和参数。
简言之,使用Cutadapt去除引物,然后修剪序列(240 f/200 r),合并12 bp重叠,去噪,并使用DADA2 进行比对。删除嵌合体,并通过IDTAXA的IdTaxa函数进行分类。
CABO-16S数据库
CABO-16S将常用的16S rRNA数据库统一整合,提供了一个用户可以轻松扩展的单一数据库,包含了数据库更新或未发表的序列。来自SILVA 138.2的389144个细菌和19213个古细菌16S rRNA序列用作CABO-16S数据库的初始框架,同时保留了来自SILVA的随机100个真核生物序列作为外群。
这些序列与来自PR 2数据库的细胞器16S rRNA基因的8540个16S rRNA序列相结合。最后,可以组合自定义序列,以最大限度地提高目标群落的分辨率;这里还添加了一组未发表的全长16S rRNA序列,这些序列是从甲烷渗漏沉积物的Sanger测序中获得的,以及一份精选的代表性seep-SRB1序列列表。
doi.org/10.1101/2024.10.23.619938
以相当的准确度注释先前未分类的ASV
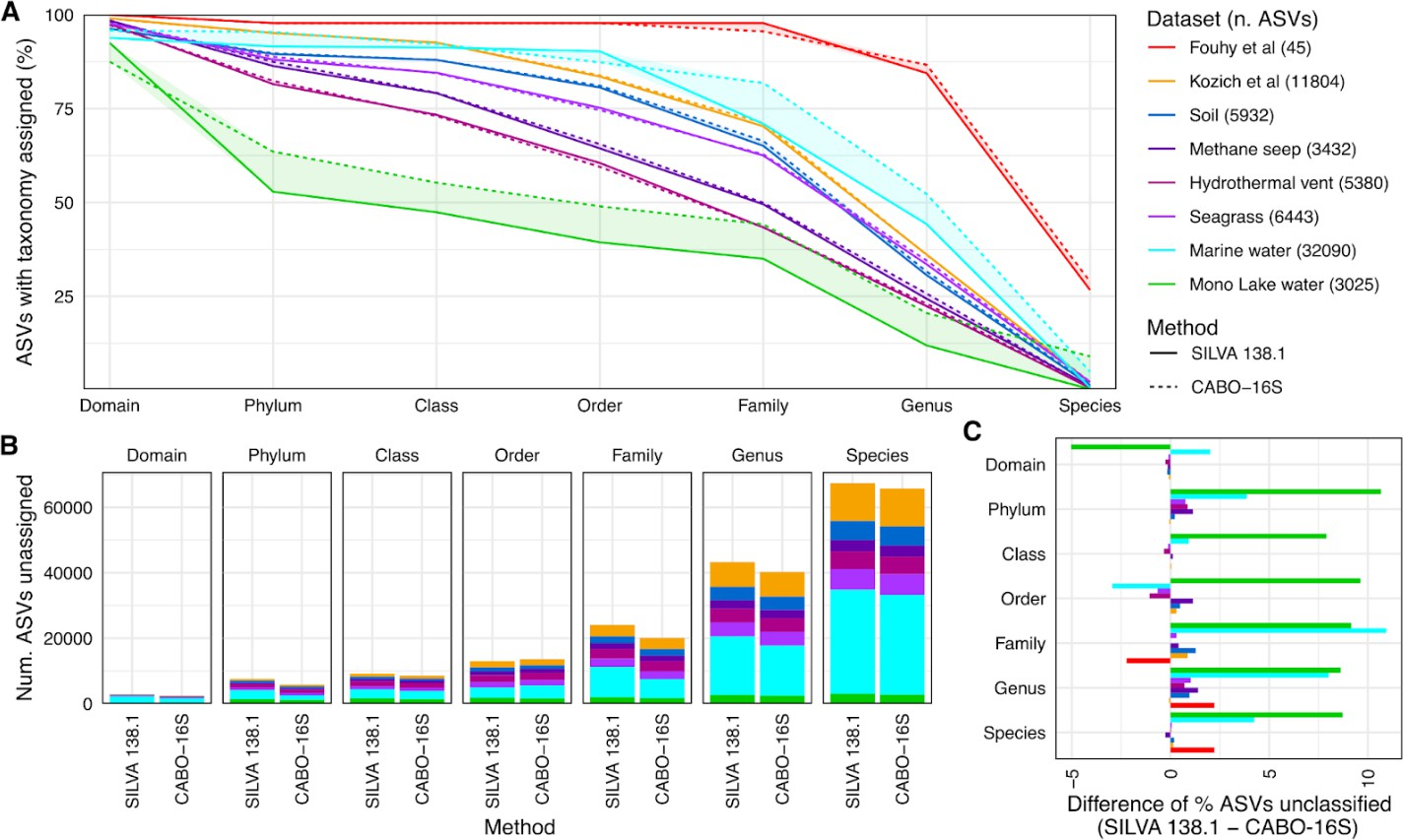
CABO-16S与SILVA 138.2比较了先前发表的代表不同系统的数据集,包括模拟群落和环境样本。该数据集包含基于模拟群落、哺乳动物肠道和住宅土壤、北方森林土壤、叶和周围沉积物、来自冷甲烷渗漏的深海沉积物、热液喷口沉积物、具有丰富浮游植物群落的海水以及最后来自以微藻Picocystis为主的封闭盆地湖泊的水的特征良好的基准集。合并数据集由64402个扩增子序列变体(ASVs)组成,单个数据集范围在45-32090个ASV之间。
CABO-16S的分类能力显著提高
在所有分类水平上接受分类分配的ASVs总数方面,CABO-16S数据库的表现优于未经修改的SILVA 138.2数据库。最大的差异是具有最多光合真核生物的数据集,如浅海草环境、海水和湖水。
例如,在海水数据集中,CABO-16S在门水平上比SILVA 138.2多分类了约10%的ASV。其他数据集差异不大,表明纳入PR2数据库的细胞器16S序列并没有对分类器继续准确预测细菌和古菌分类的能力产生有意义的影响。
唯一值得注意的例外是,SILVA 138.2中的海水数据集的分类率略高于CABO-16S,我们将其归因于PR2中的浮游植物目数比SILVA中的单一“叶绿体”标签增加。在这两个数据集中,绝大多数ASV都无法在物种水平上进行分类,尽管这可能部分是由于标记到物种水平的参考序列相对较少,特别是对于非人类环境特有的微生物。
doi.org/10.1101/2024.10.23.619938
A)SILVA 138.2(虚线)和CABO-16S(实线)在给定分类水平(x轴)上按数据集(彩色线)分类的ASV百分比。随着ASV在较高级别(如域)缺乏注释,这些线也在不断减少,根据定义,ASV在较低级别(如物种)也缺乏注释。
B)A.缺乏分类的ASV的绝对数量。
C)两个数据库之间未分类的百分比差异。阳性百分比反映CABO-16S注释的ASVs比SILVA 138.2更多,反之亦然。
CABO-16S和SILVA都揭示了分类模糊性的两种类型之间的区别,这是妨碍分类注释的主要原因。模糊性是最常考虑的精度形式,其中一个序列位于两个或多个参考分类群之间,因此不能在选定的置信阈值(本研究中为40%)下分配给单个分类群。
IDTAXA和其他类似的分类器通过将序列分类到竞争参考分类群的最低共同水平来处理此类事件,有时会在冲突的分类等级中添加‘unclassified’未分类前缀。相反,一个序列可以被确信地分配给一个单一的分类单元,但是如果参考序列在给定的等级上缺乏注释,那么在该等级上仍然可能缺乏分类学。
这种情况会影响许多未培养的谱系,例如,候选门级辐射类群candidate phyla radiation(Patescibacter门)中的SR1 科在SILVA 138.2中没有属或种分类,所有121个序列仅在科水平上进行注释。
因此,缺乏属分类的SR1 ASV不是由于分类器的不确定性,而是由于分类学的不确定性。此外,一些谱系可能同时包含这两种不确定性来源,例如,在SILVA 138.2中,Desulfosarcinacae脱硫杆菌科下有53个序列标记为种水平、676个序列标记为属水平和345个序列标记为科水平。而脱硫杆菌科的ASV缺乏属水平注释可能是由于与仅标记到科级的一组序列有密切相似性(taxonomic ambiguity 分类学模糊性)或与不同属无法区分(classifier ambiguity 分类器模糊性)。
因此,我们依照IDTAXA惯例在分类器模糊的情况下加上“unclassified_”,以及在参考序列模糊的情况下在最低分类层级上添加“unspecied_”来区分两者。
CABO-16S能够分类出几乎占一半读数的真核叶绿体
在分类序列中,CABO-16S和SILVA在大部分数据集中都产生了相似的群落组成。事实上,由于CABO-16S古菌和细菌序列的非蓝藻部分与SILVA 138.2完全相同,因此预期会有这种一致性。然而,在含有光养真核生物的数据集中(例如海草、海洋和湖泊水柱数据集),CABO-16S数据库允许对真核叶绿体进行分类,这些叶绿体占某些样本读取量的近50%(Mono Lake数据集)。
迄今为止,海水数据集的浮游植物多样性是所有数据集中最大的;这种多样性中的大部分都可以用CABO-16S进行分类标记。在Mono Lake数据集中,通过与NCBI人工对比,剩余的未分类多样性可归因于浮游植物线粒体序列。虽然目前的PR2数据库包括大约1842个线粒体序列,但绝大多数(1782个 96.7%)属于Opisthokonta,只有22个序列属于泛植物界Archaeplastida(植物和许多藻类)。
虽然并非在所有真核细胞中都发现线粒体,但我们预计未来PR2的扩展将包括更多来自植物和藻类谱系的线粒体16S,这将改善这一问题。
CABO-16S与原始SILVA 138.2的每个数据集的组成
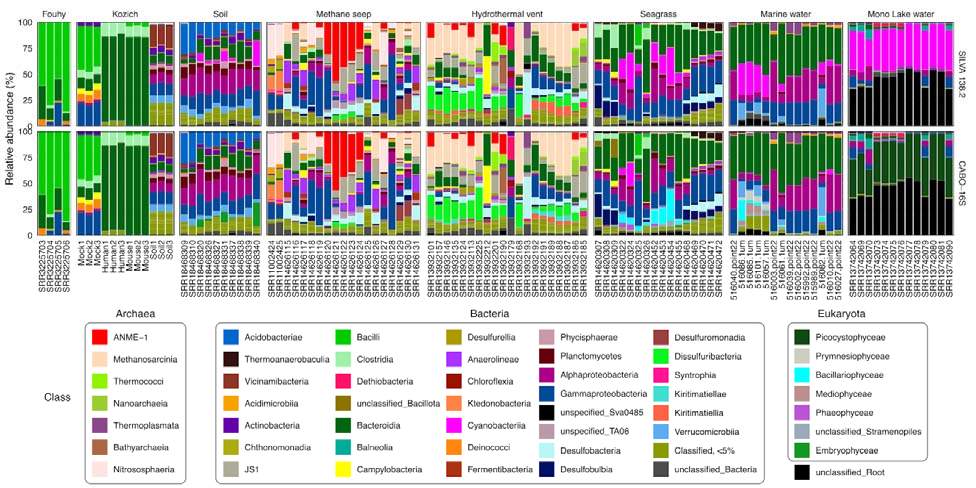
ASV被聚合到类级别(用不同颜色表示)。在样本中含量在5%以上的类别在图中展示。无法分配特定类别的ASV同样被汇总在最低注释级别。相对丰度<5%的剩余ASV被归为一类。
鉴定光合真核生物的精度更高
通过每个光养真核生物数据集对比不同ASV分类,以阐明数据库之间的差异点。对于富含真核浮游植物的沿海海水数据集,SILVA 138.2能够准确地对细菌群落进行分类,但是大部分reads(推测为真核生物)没有被分类到域水平或简单地注释为科水平的叶绿体。
扩增子序列变体序列得到进一步分类
而在CABO-16S数据集中,这些相同的质体ASV序列得到了进一步的分类。值得注意的是,序列的多样性并不总能对较低水平进行明确的分类注释,图中的分类注释显示在类或目水平上出现了许多歧义。
一些分类单元等级包括‘ _X ’后缀,这是PR2使用的中间占位符,类似于其他分类中使用的‘ Incertae Sedis ’。然而,通过质体分类获得的分类注释是有用的,因为主要的浮游植物类群(如硅藻、鞭毛藻、隐生植物等)是有区别的。
CABO-16S解析海洋数据集中的真核和细菌浮游植物
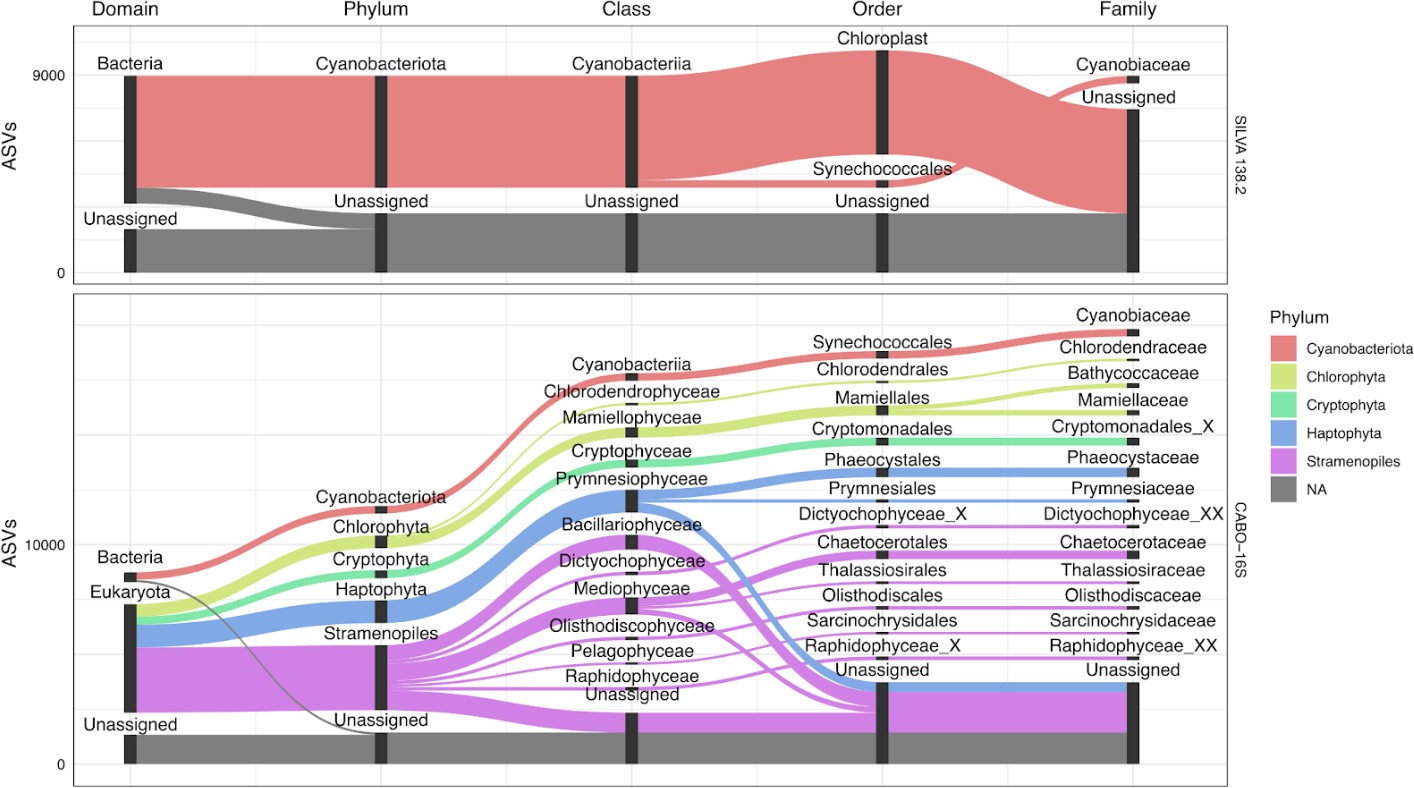
SILVA 138.2和CABO-16S的冲积图追踪了至少有100个ASV从域(最左侧)到科(最右侧)的ASV分类。对于每个层级,柱状图代表不同的分类群,每个柱状图的大小按比例反映了ASV的数量。条形图之间的空白是为了便于可视化。连接层级的流根据这些ASV的门级分类进行着色。仅显示了基于任一数据库的与蓝藻或质体序列相关的ASV。
自定义序列增加了多系进化枝的鉴定
向CABO-16S中添加自定义序列可以增加目前未纳入SILVA或PR2数据库中的物种的分类。增加了Sanger测序获得的甲烷渗漏沉积物序列和代表性SEEP-SRB1序列的精选列表。
SEEP-SRB 1是硫酸盐还原菌的多系分支,包括已知的ANME在甲烷厌氧氧化(AOM)过程中的共生性成员,如SEEP-SRB1a和SEEP-SRB1g,以及其他非共生性成员(SEEP-SRB1b、SEEP-SRB1c、SEEP-SRB1d、SEEP-SRB1e和SEEP-SRB1f)。
虽然目前在SILVA 138.2中被确定为属级进化枝,但这是这些生物的一个过于简化的分组。事实上,虽然一些成员,如SEEP-SRB1g和SEEP-SRB1c,被描述为物种水平的分支,但其他成员,如SEEP-SRB1a,更准确地描述为属水平的分支。
分辨率的提高揭示了不同SEEP-SRB 1亚组的不同分布****
进一步使SEEP-SRB分类学复杂化的是SEEP-SRB1亚群之间不对称的系统发育距离——例如,基于基因组树,SEEP-SRB1g和SEEP-SRB1a可能位于不同的序列中。虽然用分类学分类纠正系统发育距离超出了本工作的范围,但这种历史命名惯例之间的冲突在环境微生物学中很常见并且难以解决。
然而,使用精确命名的组扩展数据库提供了一种避免这些差异的方法。因此,我们将SEEP-SRB亚群的代表性序列添加为SEEP-SRB1的“种”,但将SRB1g添加为“种”,这是SILVA 138.2中与SRB1g最相似的序列的分类名称。
将这些额外的SEEP-SRB序列纳入CABO-16S数据库,将部分环境SRB1组ASVs分解为各自的亚组。在甲烷渗漏和热液喷口数据集中,分辨率的提高揭示了不同SEEP-SRB 1亚组的不同分布。在喷口数据集中,只有一部分样本含有同营养型Seep-SRB1a和非同营养型Seep-SRB1d,这一区别无法用默认的SILVA 138.2数据库解决。与SILVA 138.2相比,CABO-16S如何将SILVA中注释为SEEP-SRB1、未分类的脱硫藻科或脱硫藻的ASVs分类时,分类分配的进一步差异变得清晰。
虽然CABO-16S对整个数据集中相对较小比例的ASV进行了不同的分类,但在特定环境中,如本研究中包括的沉积热液喷口,分类差异是显著的。有趣的是,一些SILVA 138.2分类为脱硫菌科的ASV在CABO-16S的较高级别上未分类(例如,unclassified_Desulfobacterales)或不同的分类。我们将这种差异归因于IDTAXA算法中的运行间差异,因为它对每次运行随机进行kmer的子采样,因此分类置信阈值边缘的一小部分ASV在运行之间会收到不同的分类。为了支持这一观察结果,将分析纳入归类为SEEP-SRB1、、Desulfosarcinaceae或LCP-80的ASVs,结果与一些归类为CABO-16S的asv在SILVA 138.2中未分类的结果大体一致。
CABO-16S通过添加特定序列,以更高的分辨率对SEEP-SRB1进行分类。

A) SEEP-SRB1及其相关分类群在最低分类水平的相对丰度。对于甲烷渗漏和热液喷口数据集,左侧子面板显示SILVA 138.2分类,右侧子面板显示CABO-16S分类。y轴表示每个样本总读取次数的百分比。
B)冲积图显示了数据库中相同ASV序列的分类。每一列都是一个不同的等级(从科到种),用CABO-16S标记的物种等级为不同的颜色。流量高度反映asv的数量。
缩写:unc,未分类;unsp不明;sed,沉积物。请注意,我们区分了由于分类冲突(未分类)而缺乏注释与由于参考分类群的注释不完整(未指定)而缺乏注释,如上图所述。
添加自定义序列相关的挑战
虽然Seep-SRB1亚型的注释可以通过添加具有特定注释的已知序列来实现,但这些分支的分类学、层级命名系统和系统发育,进化史之间仍然存在差异。
其他小组可能需要比我们用于SEEP-SRB1的方法更复杂的方法。志贺氏菌属和埃希氏菌属是这种矛盾的象征,因为两者在进化上都有很深的重叠,但分类学继续使参考数据库层次结构复杂化。
几十年来,聚球藻等其他环境群体同样对纠正分类学和系统发育提出了挑战。对于此类群体,添加具有特定、系统发育正确层次结构的序列不太可能改善分类,因为解决歧义的LCA方法假设所有序列共享相同的层次结构。
因此,所有现有序列都需要按照所需的系统发育框架进行类似的重新分类,并需要额外的管理,以确保新的分类层次与序列相似性的兼容性。最终,纠正系统发育和分类学的可行性受到16S rRNA基因中嵌入的信号的限制,虽然基于基因组的系统发育和16S rRNA系统发育在很大程度上是一致的,但它们并不完全相同。
◮ 分类分配的错误或不一致
提高分辨率的另一个障碍是分类分配中的错误或不一致,即相似的序列具有冲突的名称;据估计,这类错误占SILVA序列的1.5-17%。
基于大多数序列被正确且一致地标记的假设,IDTAXA等方法结合了工具来识别和删除在训练期间与大多数相似命名的序列冲突的单个序列,并且也存在独立的工具。
然而,这种方法对于由许多序列代表的分类群效果最好,对于需要提高分辨率的环境谱系并不总是如此。分类器的分辨率和准确性也可以通过限制数据库只包括特定于采样栖息地的微生物来提高,正如许多动物微生物组已经成功做到的那样。
这种特定栖息地的训练集无疑是对特定系统进行集中研究的最佳方法。但是,要了解具有广泛分布的特定分类群(如SEEP-SRB)的环境背景,就需要使用诸如SILVA之类的通用数据库最大化分辨率的方法。
结语
CABO-16S成功地将来自SILVA 138.2的细菌和原始16S rRNA序列以及来自PR2数据库的细胞器16S rRNA序列与自定义选择的序列结合起来。与SILVA 138.2相比,**增加了****扩增子序列变体(ASVs)**的分类定位。
具体来说,通过添加PR2的质体序列,CABO-16S无需额外测序16S和18S引物,就能在海洋和湖泊水体中出色地鉴定光养真核生物。尽管一些16S序列,如来自植物和藻类的线粒体仍然很少,可能会影响特定环境的分类,但CABO-16S减少了未分配的光养生物的数量从而可以快速提取剩余的丰富序列。
CABO-16S也被构建为可以添加自定义序列。随着SEEP-SRB1多系分支序列的加入,我们看到热液喷口沉积物样本的分类分化增加。这有助于确定特定环境中综合征的可能性,并加深对AOM的社区的理解。尽管添加自定义序列必须谨慎进行,但考虑到SILVA中未指定序列的数量以及将多系分支限制在当前分类结构的难度,CABO-16S的这一功能使用户可以自由定制16S分类,并可能增加对特定环境的理解。
最后,CABO-16S提供了一个框架,可以随着SILVA和PR2数据库未来版本的发布而轻松更新。R脚本和工作区可在github.com/emelissa3/CABO-16S上获得。自定义序列和其他东西永久托管在Figshare(doi.org/10.6084/m9.figshare.27288090)。
参考文献:
CABO-16S – A Combined Archaea, Bacteria, Organelle 16S database for amplicon analysis of prokaryotes and eukaryotes in environmental samples.
Eryn M.Eitel, Daniel Utter, Stephanie Connon, Victoria J. Orphan, Ranjani MuralibioRxiv 2024.10.23.619938


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国