

导读
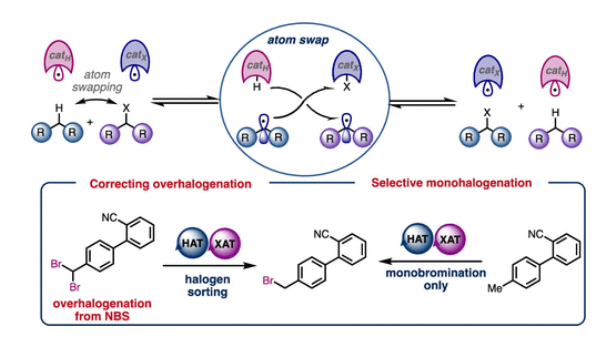
近日,苏黎世联邦理工学院Bill Morandi团队基于自由基交换策略开发了一种C−H/C−X键的复分解反应(Metathesis,即“交换舞伴”),其中氢原子与卤原子在分子间通过可逆的氢原子转移(HAT)和卤原子转移(XAT)进行交换,从而实现温和的C−H键卤化反应。该过程的可逆性使得多卤代产物可选择性脱卤,生成单卤代产物。利用此可逆性,有机卤化物污染物亦可作为C−H键卤化的卤源。总体而言,该策略证实了在C−H键官能团化中引入可逆复分解理念,可为合成策略提供互补性优势。文章链接DOI:10.1021/jacs.5c04754
(图片来源:J. Am. Chem. Soc.)
正文
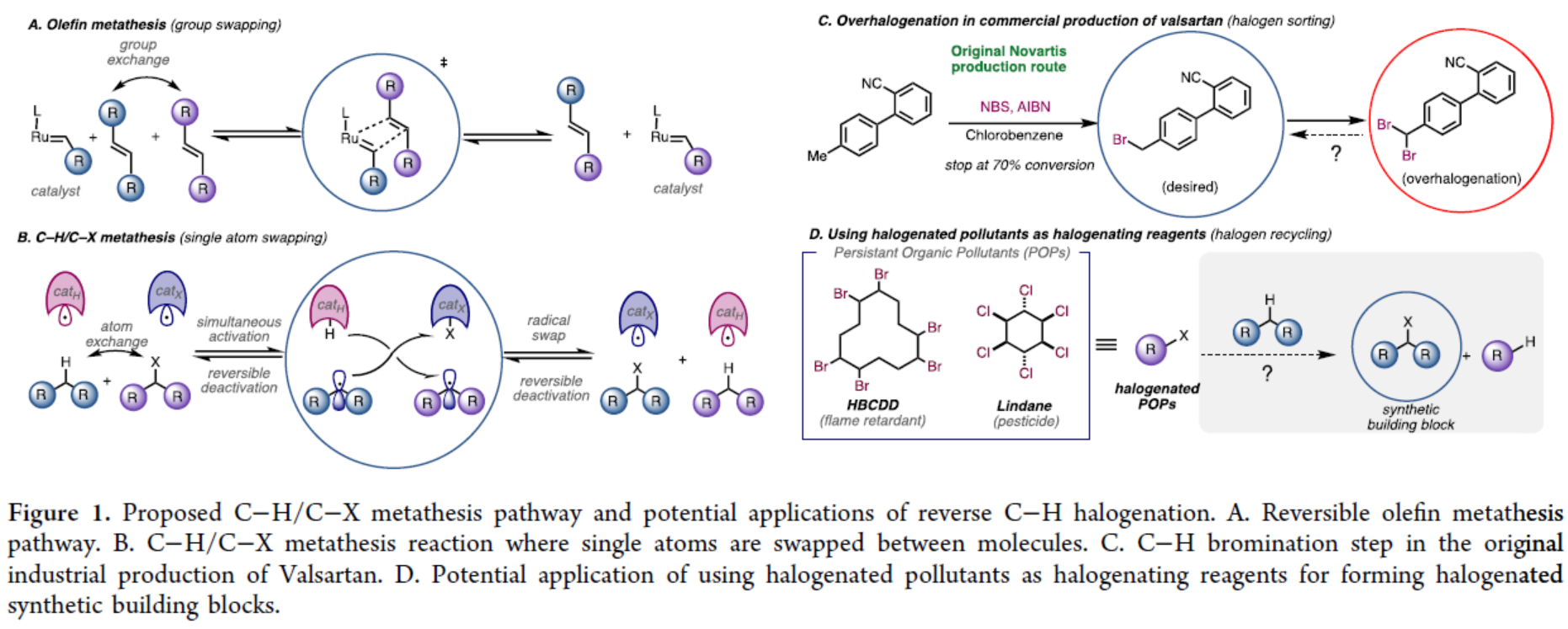
复分解反应代表了一种温和且具有互补性的合成策略,可用于构建复杂分子与聚合物。该反应能够通过同一催化体系进行正向或反向反应,从而构建或解构化学键,并能实现传统反应无法完成的分子片段的可逆重组。烯烃复分解反应(Figure 1A)是可逆复分解反应中最具代表性的案例之一,在生物质增值、商品和精细化学合成、废物回收利用、超分子组装和可循环聚合物的合成等不同领域都有突破性的应用。基于复分解策略,Morandi等团队已开发出两种不同活性官能团间的催化型官能团化复分解反应。然而,这些新方法以及上述成熟应用的普适性仍存在明显局限,即两种反应物均需预先引入活性的官能团。另一方面,C−H键官能团化可利用广泛存在的C−H键直接构建复杂产物,且无需进行预官能团化。因此,将可逆复分解策略应用于C−H键官能团化,有望融合这两大前沿概念,从而为合成化学开辟全新前景。然而,到目前为止,此类方法仍然没有得到充分的探索。在利用C−H键进行可逆复分解反应的罕见案例中,最显著的是C(sp2)−H键至C(sp2)−硼化/碘化反应,其自校正能力可产生独特的选择性。除上述方法外,C−H复分解反应在工业领域的重要应用是甲苯的C−H/C−Me复分解反应,可用于制备二甲苯与苯。基于这些先例,将非活化的C(sp3)−H键纳入复分解策略具有特殊吸引力,既可拓展C−H官能团化的能力,又可催生新类型的复分解反应(Figure 1B)。基于C-H键复分解的概念(Figure 1C与Figure 1D),Morandi团队认为苄位C(sp3)−H键卤化反应兼具吸引力,且可对传统方法形成互补。尽管现代试剂已实现非活化C(sp3)−H键的卤化,但苄位C(sp3)−H键的卤化仍主要依赖经典试剂,如卤素单质(Br2或Cl2)或AIBN/NBS或AIBN/NCS复合体系。然而,这些试剂易导致过度卤化,且需针对特定底物单独优化反应体系。同时,使用这些方法无法实现逆反应(即去卤化反应),且需要不同条件,这些条件通常涉及有毒锡试剂,尽管现代方法提供了替代方案。“过度卤化”是另一个与经典方法相关的常见问题,在此情况下,复分解方法可通过逆反应再生所需的单卤代产物。 欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
(图片来源:J. Am. Chem. Soc.)
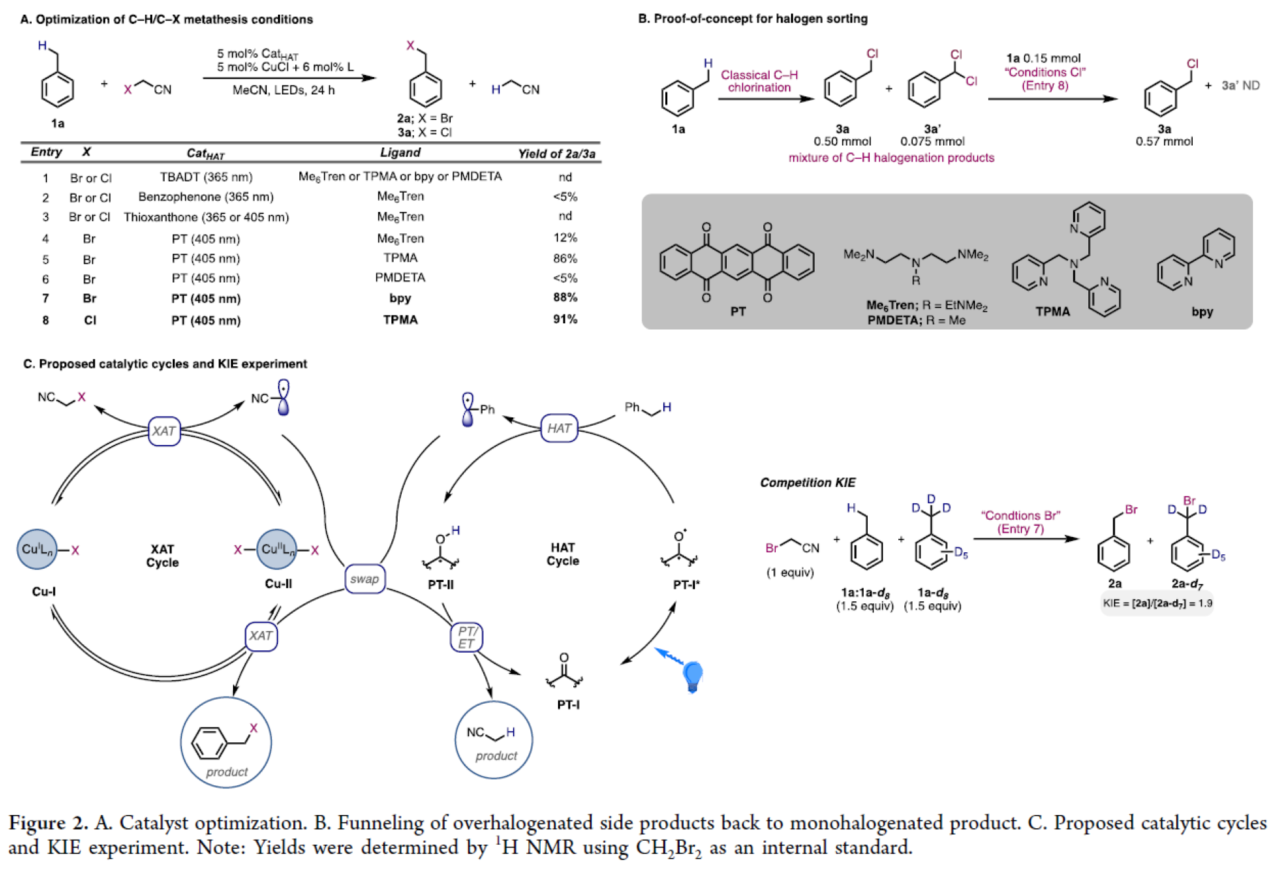
首先,作者以甲苯1a与溴乙腈(作为溴源)作为模型底物,进行了相关反应条件的筛选(Figure 2A)。当以5,7,12,14-并五苯四酮(PT,5 mol %)作为HAT试剂,CuCl(5 mol %)作为金属催化剂,bpy(6 mol %)作为配体,405 nm LEDs作为光源,在MeCN溶剂中室温反应24 h,可以88%的收率得到苄基溴产物2a。同时,当以氯乙腈作为氯源,TMPA(6 mol %)作为配体,同样可以91%的收率得到苄基氯产物3a。
其次,为测试体系在选择性卤素分选方面的效能,将单卤代化合物3a(0.50 mmol)、双卤代化合物3a′(0.075 mmol)和非卤代化合物1a(0.15 mmol)的混合物(比例∼7:1)置于优化反应条件下(Figure 2B)。在3a′完全转化后,可获得产物3a(0.57 mmol),收率为86%。除了其机理价值外,该实验还表明利用卤素分选可将多卤代异构体混合物转化为单一产物的可能性。
此外,作者提出了一种可能的HAT和XAT协同催化机理(Figure 2C)。该HAT催化剂在动力学控制下进行(与KIE实验相符,Figure 2C),并在正向反应中选择性断裂空间位阻可及的最富电子C−H键。在反向反应中,其倾向于通过质子-电子协同转移到缺电子自由基来形成缺电子的C−H键。而XAT催化剂可同时活化缺电子与富电子C−X键,但对缺电子C−X键的活化速率更快。随后,缺电子的Cu-II中间体更易与富电子自由基结合(而非缺电子的氰甲基自由基)。尽管所有C−X键的活化过程均完全可逆,但由于HAT循环中存在其他不可逆或慢速步骤,苄位C−X/C−H键的交换速率极慢。
(图片来源:J. Am. Chem. Soc.)
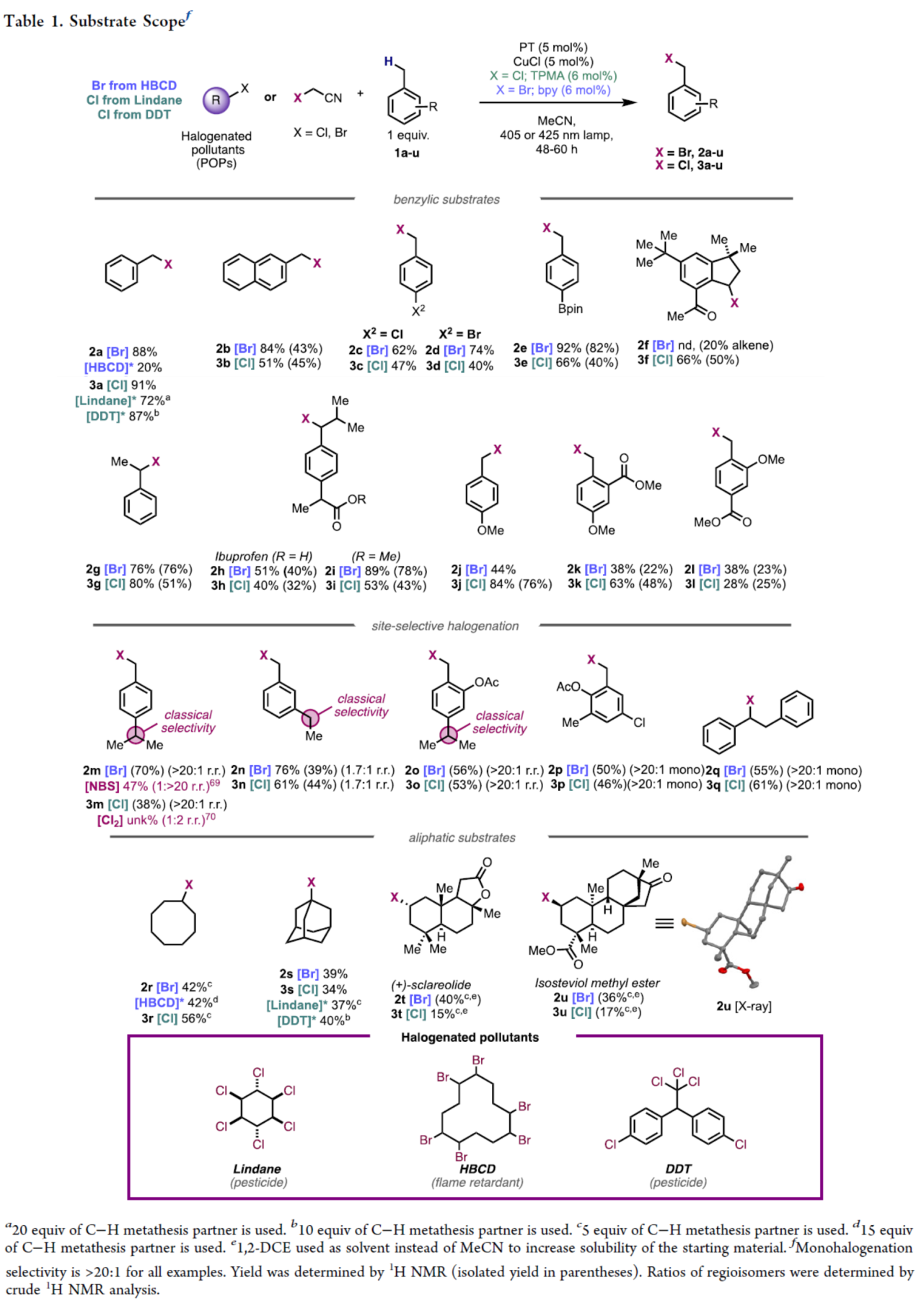
在获得上述最佳反应条件后,作者对底物的范围进行了扩展(Table 1)。首先,简单的苄基和萘基衍生物,均可顺利进行多种卤化反应,获得相应的溴化产物2a-2e(收率为62-92%)和3a-3e(收率为40-91%)。二级C−H键(1f−1i)在溴化和氯化反应中也具有良好的产率,尽管环状体系进行溴化会导致脱饱和形成苯乙烯(2f)。未受保护的药物分子布洛芬,在2小时和3小时内提供了所需的产物,对更富电子的苄基C-H键具有完全的选择性,并且没有竞争性的脱羧反应。含有甲酯保护的布洛芬,显著提高了溴化产物2i的收率。对位取代的供电子基团在氯化反应中表现良好(产物3j),但溴化产物2j的产率较低且不稳定难以分离。邻位取代底物(1k与1l)在溴化与氯化反应中均收率下降,并残留大量未反应原料。当芳环任意位置存在强吸电子基团(如Ar−NO2/−CF3)或缺电子杂环(如吡啶类)时,反应完全不能发生,表明此类C−H键缺电子性过高而无法被活化。
其次,采用大位阻PT催化剂突破了传统C−H键卤化的选择性限制,实现了空间位阻最小C−H键(而非键能更弱的C−H键)的优先官能团化。对异丙基氯苄(p-Cymene)对甲基具有完全的选择性,这与NBS或Cl2的选择性形成鲜明对比。对于底物1n,甲基甚至可超越苄位仲碳实现选择性卤化(1.7:1选择性),逆转了现代方法的常规趋势。
此外,对于非活化的脂肪族C-H键,当使用一当量的C-H起始底物时,产率较低。因此,使用过量的C-H底物,环辛烷可以良好的收率得到产物2r和3r,金刚烷也可以良好的收率得到1-卤代金刚烷产物2s和3s,具有完全选择性。使用天然产物(+)-香紫苏内酯和异甜菊醇甲酯(isosteviol methyl ester),可获得具有完全区域选择性的卤化产物2t/3t和2u/3u。值得注意的是,使用DDT或Lindane作为限制试剂,甲苯(1a)可以高产率进行氯化得到产物3a,而金刚烷(1s)等脂肪族底物则提供中等收率的产物3s。使用HBCD作为溴源,环辛烷也可以良好的收率进行卤化反应。
(图片来源:J. Am. Chem. Soc.)
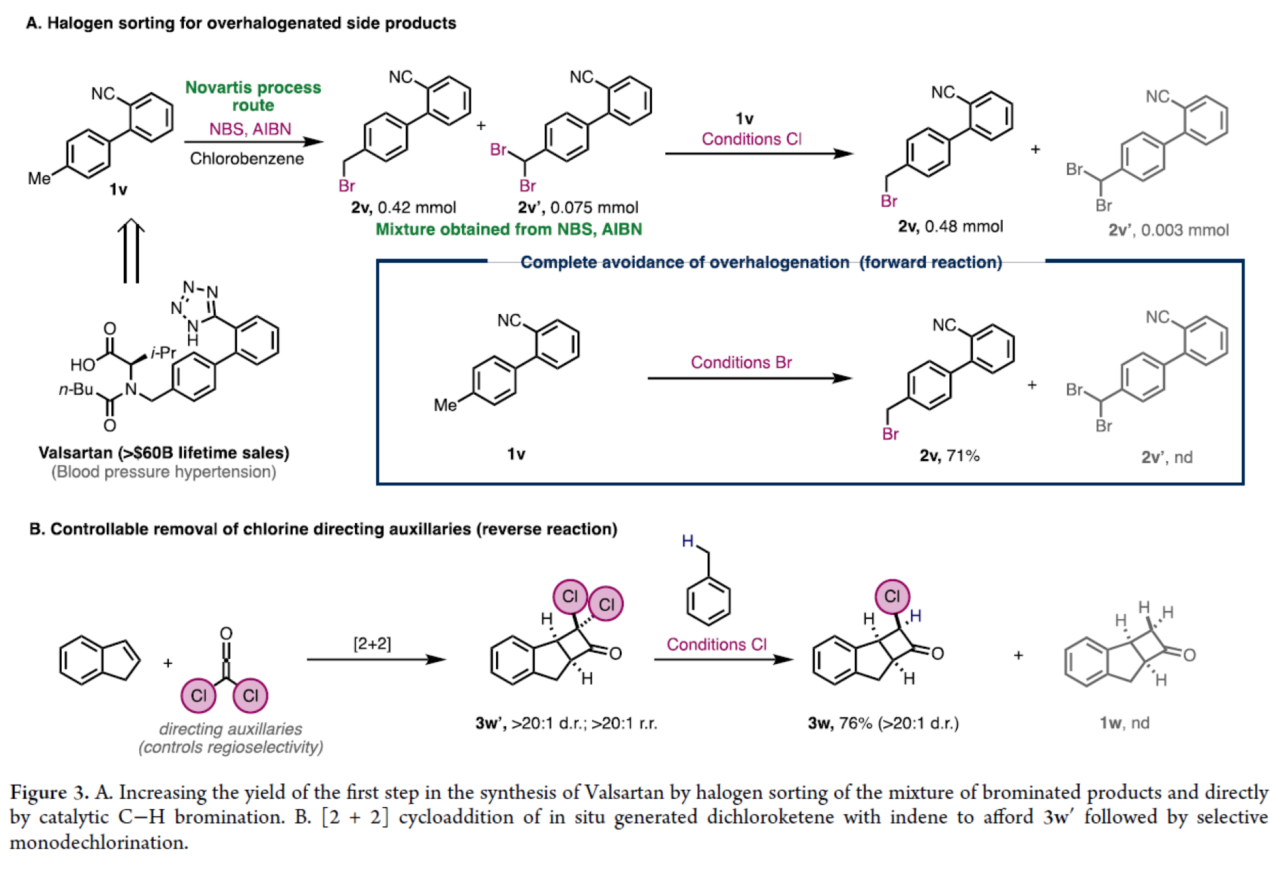
最后,作者利用逆反应和可逆卤化进行卤素分选(halogen sorting)反应(Figure 3)。在诺华公司报道的缬沙坦的原生产工艺中,1v的C-H溴化反应需在氯苯溶剂中使用NBS 和AIBN在100 ℃下进行。为防止因过度卤化导致产物损失,需严格监控反应进程并在转化率达70%时终止。随后,蒸除氯苯,经溶剂洗涤移除过度卤化副产物2v′,再补加NBS对剩余原料进行二次溴化。采用本方法,在80%转化率时中止反应,并将混合物置于催化氯代条件下处理,此时副产物2v′可转化为目标产物2v。作为替代方案,在标准催化溴化条件下,1v可直接专一性的生成2v,收率达71%。此外,利用[2+2]二氯乙烯酮-烯烃环加成反应的产物3w′,其发生高选择性单脱氯反应,以高产率生成单一的非对映异构体3w。Cu-I对缺电子程度最高的卤素表现出高化学选择性,同时苄氯(3a)的形成可能抑制了完全脱氯过程,从而实现可控的单脱氯转化。事实上,使用经典的Zn/H+条件形成相同的多功能合成中间体3w的尝试,只提供了3w、3w′和1w的混合物。
(图片来源:J. Am. Chem. Soc.)
总结
苏黎世联邦理工学院Bill Morandi团队基于复分解反应概念,开发了一种新型C−H键官能团化催化的策略。该C−H复分解反应利用广泛存在的C−H键,在两种分子间交换氢原子(H)与卤素原子(X),高效合成结构多样的卤代产物。此外,反应的模块化设计特性有助于拓展应用场景,实现其他官能团在C−H复分解体系中的高效整合。
文献详情:
C−H Functionalization via Single Atom Metathesis of C−H and C−X Bonds.
Tanner C. Jankins, Barnabé Berger, Françoise A. Aouane, Sergio Barbeira-Arán, Christophe Didier, Bettina Hürlimann, Claudius Zimmer, Bill Morandi*.
J. Am. Chem. Soc. 2025
DOI:10.1021/jacs.5c04754
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国